-
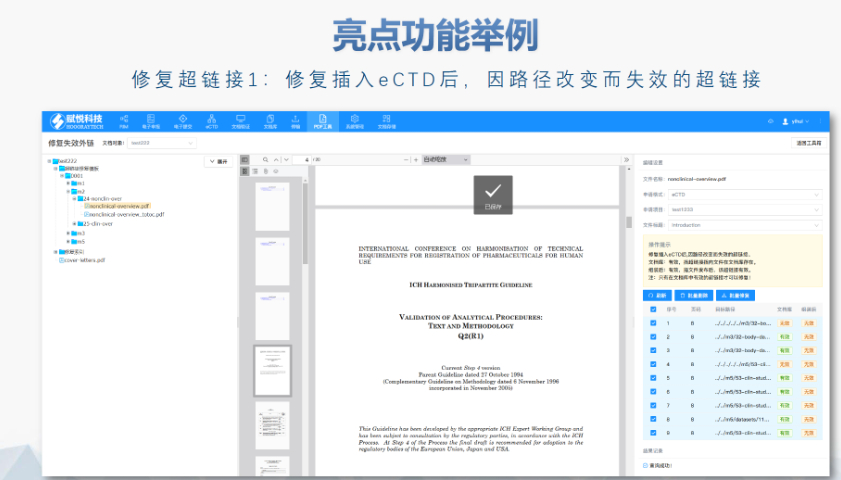 江苏国内注册eCTD报价12.18
江苏国内注册eCTD报价12.18ANDA一般不需要提供临床前(动物)和临床(人体)数据来证明其安全性和youxiao性(即免毒理和临床),作为替代,申请人必须合理证明其产品与原研YAO相比是shengwu等效的。按照《联邦食品、YA...

-
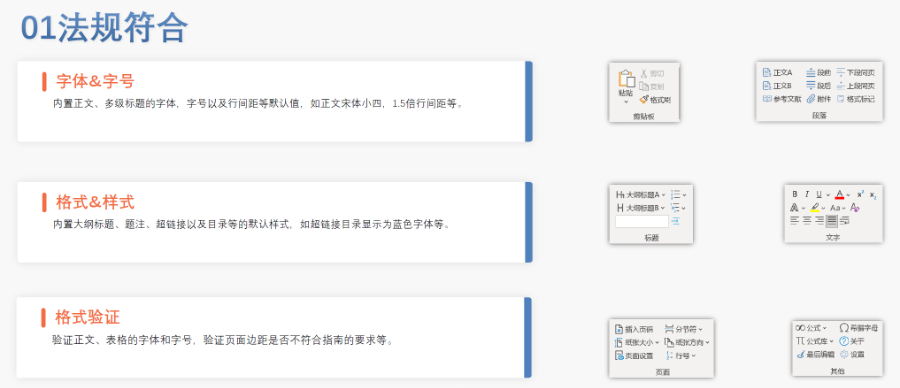 山东ANDAeCTD服务价格12.17
山东ANDAeCTD服务价格12.17ANDA一般不需要提供临床前(动物)和临床(人体)数据来证明其安全性和youxiao性(即免毒理和临床),作为替代,申请人必须合理证明其产品与原研YAO相比是shengwu等效的。按照《联邦食品、YA...
-
 工业园区eCTD报价12.15
工业园区eCTD报价12.15生命周期管理与变更递交eCTD支持全生命周期管理,申请人需通过序列更(Sequence)反映yao品变更信息。例如CEP的更需提交“变更说明表”,对比已批准和拟修改内容,并附修订版技术文档...
-
 上海eCTD发布系统12.14
上海eCTD发布系统12.14文件生命周期管理:eCTD支持文件替换(Replace)、删除(Delete)等操作,而非增文件。例如,更临床研究方案时需用Replace操作覆盖旧版本。基线提交(BaselineSubmission...
-
 闵行区仿制药eCTD使用09.22
闵行区仿制药eCTD使用09.22eCTD:FDA于2023年启动eCTD,2024年9月正式接收申请,计划2029年完成全过渡。RPS标准替代XML,支持双向通信和跨申请文件复用,例如同一StudyID可在IND和NDA...
-
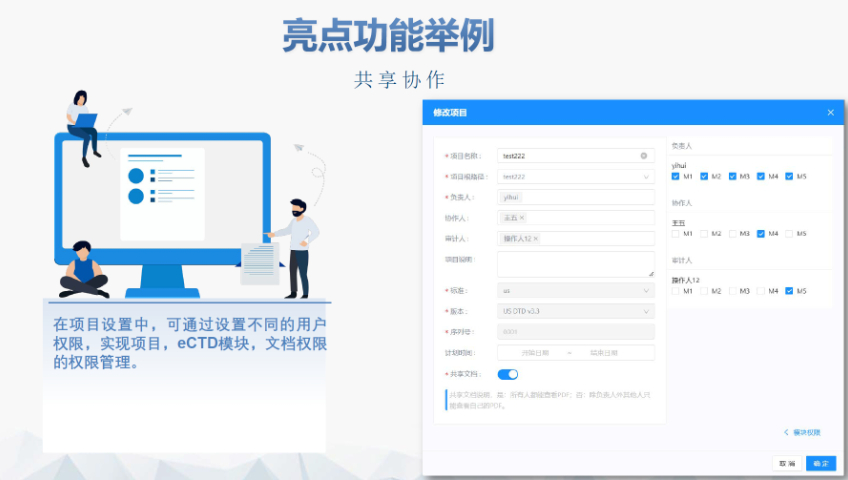 浦东新区国内注册eCTD使用05.27
浦东新区国内注册eCTD使用05.27eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF...
-
 闵行区生物制品eCTD服务价格04.26
闵行区生物制品eCTD服务价格04.26法规文档管理系统 协同共享 RDMS可以让跨区域、跨部门协同真正成为1+1>2的 工作。让频繁的文档共享传输,版本管理,生命周 期审批都变得轻松简单 安全合规 通过详细的审计追踪、电子签名、权限管理、...
-
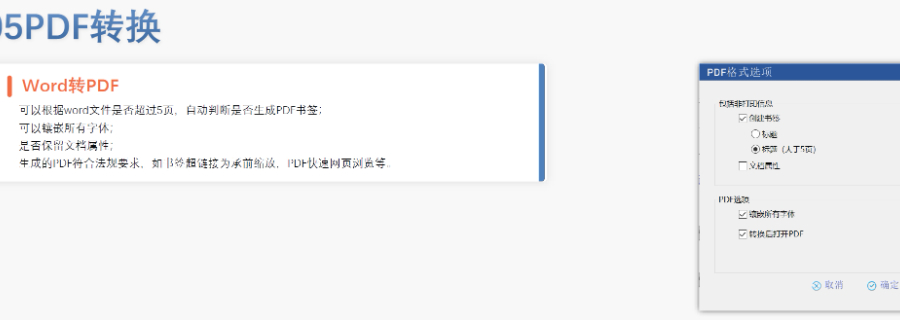 太仓中国eCTD名称04.23
太仓中国eCTD名称04.23美国eCTD的强制实施时间与范围:美国自2017年5月5日起要求药申请(NDA)、仿制药申请(ANDA)和生物制品许可申请(BLA)必须通过eCTD格式提交,2018年5月5日进一步扩展至临床试验申请...
-
工业园区NDAeCTD服务价格04.20
美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效...
-
 工业园区国产eCTD递交04.17
工业园区国产eCTD递交04.17eCTD在欧盟药品监管中的历史背景:欧盟eCTD(电子通用技术文档)的发展始于对临床试验和药品审评流程标准化的需求。2001年,欧盟引入《临床试验指令》(CTD)作为统一的法律框架,但其分散的成员国申...
-
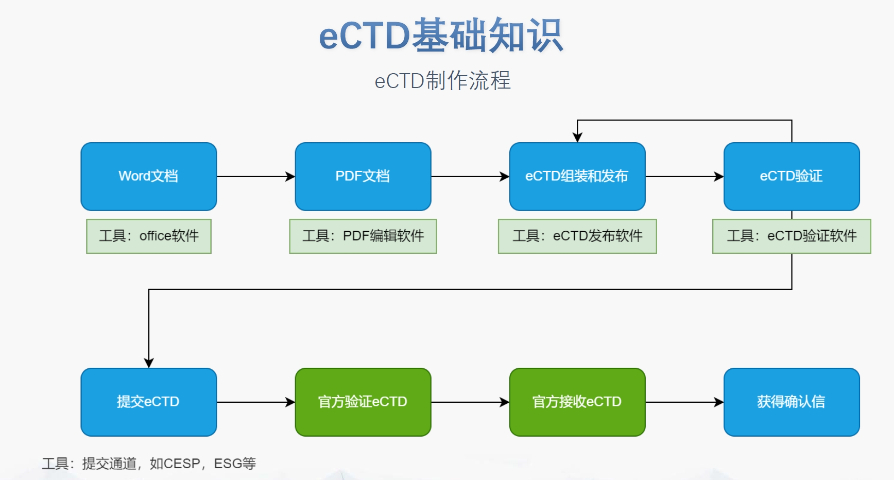 浦东新区生物制品eCTD供应商04.14
浦东新区生物制品eCTD供应商04.14电子签章与传输安全 文件需经AES-256加密后刻录至不可擦写光盘,并附MD5校验码。光盘损坏或病毒污染将触发重递交流程,原载体按销毁程序处理。 审评与核查协同 自2018年起,FDA要求提交两套光...
-
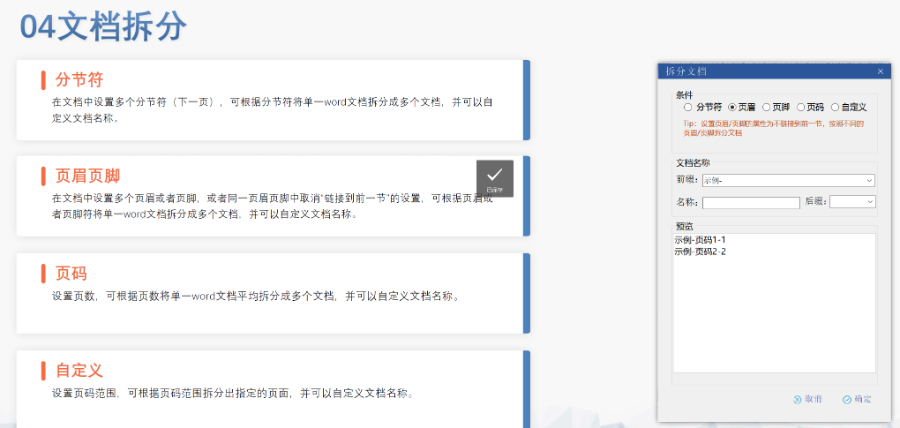 苏州ANDAeCTD性价比高04.11
苏州ANDAeCTD性价比高04.11欧洲药品管理局:集中审评程序由欧洲药品管理局(European Medicines Agency, EMA)负责协调。 人用药品委员会:人用药品委员会(Committee for Medicinal ...